醫學院徐大春團隊聯合生命科學與技術學院魏珂團隊在《循環》上發表擴張型心肌病機製研究成果
來源:生命科學與技術學院
時間:2023-04-18 瀏覽😚☂️:
擴張型心肌病(DCM)是心衰的主要發病原因之一,主要表現為心室擴大和心臟收縮功能下降👎🏿。截至2015年,全球心肌病患者約250萬例,10年內增長27%🔋。目前已經確定部分心肌結構基因突變可以誘發DCM,但仍有超過50%的病例不是由基因突變導致;也有研究表明擴心病患者心肌細胞凋亡增加、能量代謝異常🧖🏽♀️、慢性氧化應激等因素可能與疾病進展相關。多數DCM患者發展為終末期心衰🫵🏼,缺乏有效的治療藥物,2年病死率達40%,已經造成了巨大的醫療衛生負擔。
4月14日,恒达平台附屬第十人民醫院徐大春課題組🌜、恒达平台生命科學與技術學院魏珂課題組在國際心血管領域權威期刊《循環》(Circulation)在線發表了題為“Jmjd4 facilitates Pkm2 degradation in cardiomyocytes and is protective against dilated cardiomyopathy” 的研究論文👈🏼🏊🏿♂️。該研究確定了Jmjd4通過羥基化丙酮酸激酶Pkm2🦠,促進其通過分子伴侶介導的自噬途徑(CMA)降解,在維持心肌細胞代謝穩態中具有重要功能,並表明靶向Jmjd4和Pkm2可能具有治療DCM以及其他代謝功能障礙心臟病的前景。

首先,研究人員分析了人類擴張型心肌病、小鼠心肌肥厚以及心梗組織中Jmjd4的表達情況✝️,發現Jmjd4在心臟疾病中表達明顯上調,表明Jmjd4很可能是心衰進展過程中的重要效應蛋白。隨後研究者構建了可誘導心肌特異性Jmjd4敲除小鼠,發現Jmjd4在成年心肌細胞中被誘導敲除後,小鼠自發出現擴張型心肌病表型,表現為心室擴張,伴隨嚴重的病理性重塑🦸🏽♀️,並迅速進展為心衰,死亡率極高。研究者對Jmjd4條件性敲除小鼠的心肌組織和體外敲減Jmjd4的心肌細胞進行了全轉錄組測序,結果發現體內🧑⚕️、體外敲減Jmjd4後的表達下降的基因均富集在線粒體代謝相關的通路上🤲🏼,而功能性研究發現Jmjd4缺失的確導致線粒體氧化呼吸水平降低。代謝組學分析發現,Jmjd4敲除心肌細胞的糖酵解過程中丙酮酸上遊底物積累,而下遊產物濃度降低🔕,提示丙酮酸代謝異常🎽。隨後🍖,研究者通過免疫共沉澱聯合質譜技術鑒定了心肌細胞中與Jmjd4相互作用的蛋白🦄🧶,發現Jmjd4與生成丙酮酸的代謝酶丙酮酸激酶Pkm2相互作用🫄🏽。為了闡述Jmjd4是否通過調節Pkm2影響心肌細胞代謝,研究者首先檢測了Jmjd4敲減(除)情況下Pkm2的轉錄本和蛋白表達量,結果發現Jmjd4敲減(除)對Pkm2的轉錄本水平沒有影響,蛋白表達量顯著升高。相應地,過表達Jmjd4使Pkm2蛋白水平顯著降低🥈。這提示Jmjd4很可能通過翻譯後機製影響Pkm2的表達。隨後研究者使用蛋白降解系統相關的一系列抑製劑及激動劑(MG132、CHX、QX77等),及免疫共沉澱等實驗,進一步確認了Jmjd4通過分子伴侶(Hsp70)介導的自噬途徑(CMA)促進Pkm2的降解🔼。
為了進一步闡明Jmjd4促進Pkm2降解的機製👬🏼,研究者通過質譜技術確定了Pkm2的Jmjd4羥基化修飾位點(K66),並通過實驗證明該位點的突變體K66R不能結合降解過程的核心分子Hsp70👨🏿⚖️。為解除積累的低酶活Pkm2對心肌細胞代謝的阻滯🧳,研究者使用Pkm2變構激動劑TEPP-46成功治療挽救了Jmjd4條件性敲除小鼠的擴心病表型。由於Pkm2在多種心臟疾病中積累,研究者在壓力負荷誘導的小鼠DCM模型中使用該Pkm2激動劑◻️,發現其同樣可以部分挽救DCM表型👼🏽,表明Pkm2很可能是心臟代謝幹預治療的普適性靶點。
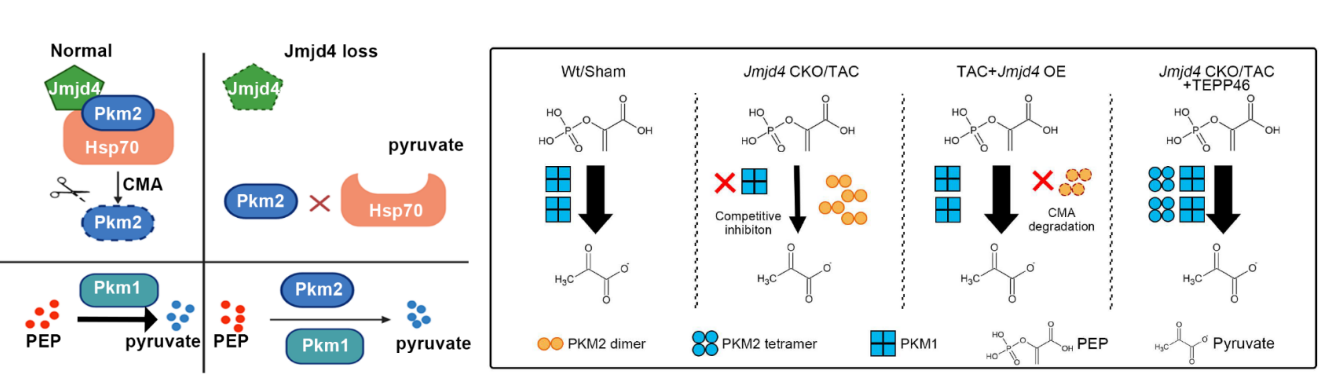
綜上所述,該研究揭示了Jmjd4通過伴侶介導的自噬調節Pkm2的降解🙂↕️,在維持心肌細胞和心臟功能的代謝穩態中起著至關重要的作用⚖️🧎🏻,這為擴張型心肌病的幹預提供了新的靶點。
該研究由徐大春教授和魏珂教授共同指導完成。恒达平台醫學院博士生唐巖松👩🏽🔧、生命科學與技術學院博士後馮夢穎為論文共同第一作者🪩。該研究得到了科技部重點研發計劃、國家自然科學基金等多項基金和教育部“細胞幹性與命運編輯”前沿科學中心的支持。
論文鏈接🧵:https://www.ahajournals.org/doi/abs/10.1161/CIRCULATIONAHA.123.064121#d40659391e203



